Sample swap of my genome from Dante Labs ?
When I learned in 2018 that Dante Labs was promoting their €169 30X whole genome sequencing (WGS) I immediately jumped on it. I have been working with Next Generation Sequencing data since I joined Illumina Inc in 2009 as a bioinformatics scientist. Recently, at the Inova Translational Medicine Institute, I published a few papers (e.g. this, this and this) on de novo mutations. Naturally, it would be interesting to look at my own de novo mutations.
The price for WGS has dropped significantly in the last 10 years but it is still quite expensive for healthy consumers who are simply “curious” about their genomes. Dante Labs’ promotional price was good enough for me to purchase it.
My parents live in Hong Kong and they had some trouble getting the right DHL label to ship the data back but after quite a few rounds with the Dante Labs customer services they got the right label and the samples were shipped!
Dante Labs has generously shared all the raw data and the variant files, providing links to download them. These include the FASTQs, BAM file, small variants, and Structural variants files. I plan on writing another entry on these so please stay tuned.
In this blog post, I am going to share the three ways that I checked my genome (a more technical blog post including scripts will come later).
TL; DR
The reasons why I think my sample was swapped can be summarized in my tweet earlier. I will go into slightly more details in this blog.
Comparison with 23andme data
My parents and I all have done 23andme so after performing some QC on the FASTQ files and looking at het/hom, ts/tv on SNVs, I investigated the genotype concordance rates between the 2 platforms in each of us.
I followed this article from bcftools to convert raw 23andme files to bcf. I used Singularity to pull the bcftools image v1.9 from biocontainers. After that, I used bcftools gtcheck to compare the converted bcf file from 23andme with the filtered.snp.vcf.gz from Dante Labs. Note that filtered.snp.vcf.gz is not a gvcf file, meaning that it does not keep track of the homozygous reference or no-call sites, however for our purpose on checking for sample matches this is sufficient.
The discordant rate for both my parents’ variants’ are ~0.08% but it is 40.5% for my genome:

Ancestry estimation
I have always wanted to try out deepvariant so I called small variants with Best Practices and generated gvcf files for each of us. And then merged the gvcfs for my family.
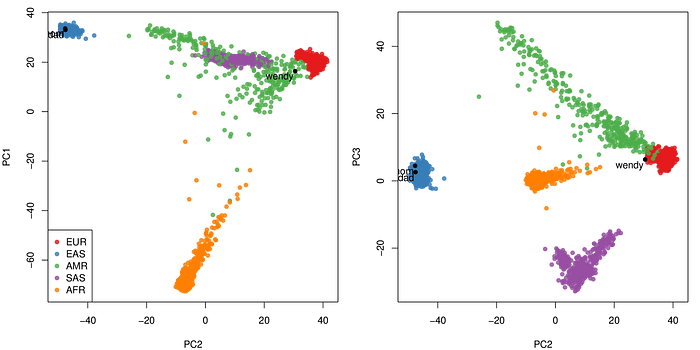
I did a quick ancestry estimation by projecting our genomes to the 1,000 genome data set, using the AKT from Illumina. It generated the figure I showed in my tweet. My family is from southern China, and as you can see in the plot. My parents’ genomes are well within the EAS (East Asian cluster), but mine is far awy from them near the EUR (European cluster).
Mendelian Inheritance Error
At this point, I am very convinced that there is a sample swap for my genome but I thought I might as well set up the analysis to look at the Mendelian Inheritance Error (MIE) anyway. I used RTG Tools to annotate my family’s merged vcf
docker run \
-v "/data":"/data" \
realtimegenomics/rtg-tools:3.10.1 mendelian \
-i /data/genomes/deepvariant/output/deepvariant.wongFamily.vcf.gz \
-o /data/genomes/deepvariant/output/deepvariant.wongFamily.annotated.vcf.gz \
--pedigree=/data/genomes/danteLabs/wong.ped \
-t /data/genomes/rtg/reference/hs37d5.sdf \
| tee /data/genomes/rtg/logs/deepvariant.wong_rtg_output.txtAnd no surprise,
2504655/7001603 (35.77%) records contained a violation of Mendelian constraintsGender Check
This is just a quick way to check to see if the sample matches with my gender. One easy way to do this is to check out the coverage in the X and Y chromosomes using samtools idxstats. For example, to get the
singularity exec -B /data/ ~/singularity_containers/samtools-v1.7.0_cv4.simg samtools idxstats <mybamfile>I get a tab-delimited output with reference sequence name, sequence length, number of mapped read-segments, and number of unmapped read-segments. The following is what I get for X and Y chromosome
X 155270560 34059561 245648
Y 59373566 185698 25220If I multiply the third column (number of mapped read segments) by 150 (read length is 150 bp), then divide by the second column (sequence length), I get the mean coverage is ~ 32.9X for the X chromosome and 0.47X for the Y chromosome. This indicates to me that the sample is indeed a female (diploid X), or you will see roughly the same coverage for X and Y chromosomes (haploid X and haploid Y).
Final Thoughts
I guess sample swap happens, the question is as the consumer how can you tell if the genome you received is indeed your genome? Of the methods I described above I guess perhaps the first thing to check is your ancestry, there are a few websites that offer such service for free, depending on the format of your genotype data. To name a few, you could try:
Secondly, if you have your genotyping done at a company, and you are able to get the raw data from them, comparing the genotypes between the 2 should also be very helpful. I will share my scripts on GitHub in my future blog posts.
Disclosure: Bear in mind that some of the links in this post are affiliate links and if you go through them to make a purchase I will earn a commission. Keep in mind that I link these companies and their products because of their quality and not because of the commission I receive from your purchases. The decision is yours, and whether or not you decide to buy something is completely up to you.